Research Articles
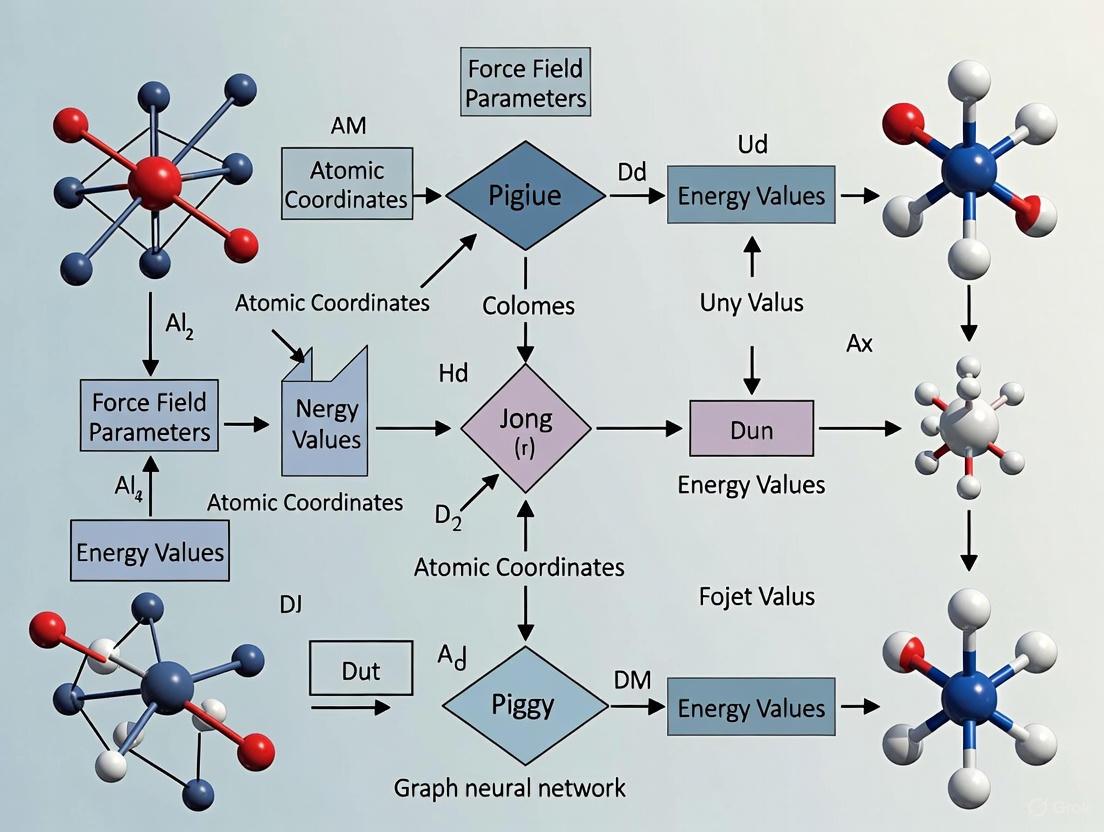
Predicting Molecular Mechanics Parameters with Graph Neural Networks: A New Paradigm for Force Field Development
This article explores the transformative role of Graph Neural Networks (GNNs) in predicting Molecular Mechanics (MM) force field parameters, a critical task for accurate and efficient molecular dynamics simulations in...
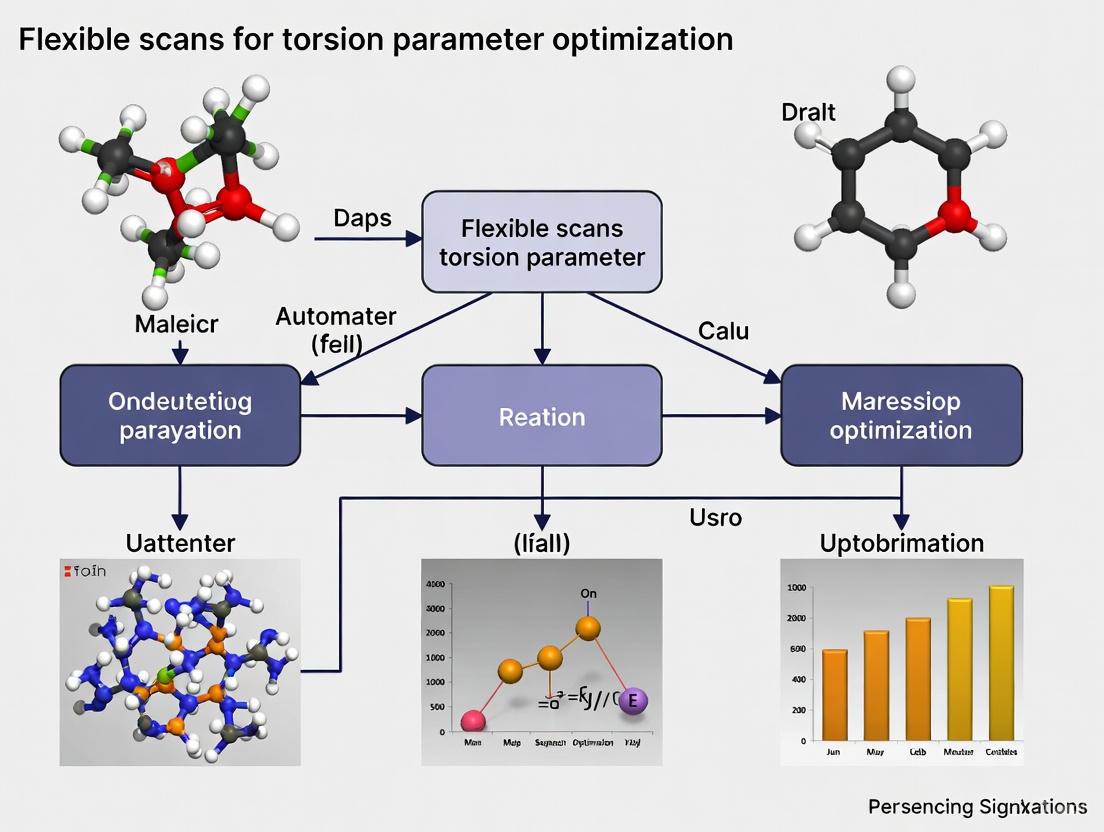
Advanced Flexible Scanning for Torsion Parameter Optimization in Biomolecular Force Fields
This article provides a comprehensive guide for researchers and drug development professionals on performing flexible scans to optimize torsion parameters in molecular simulations.
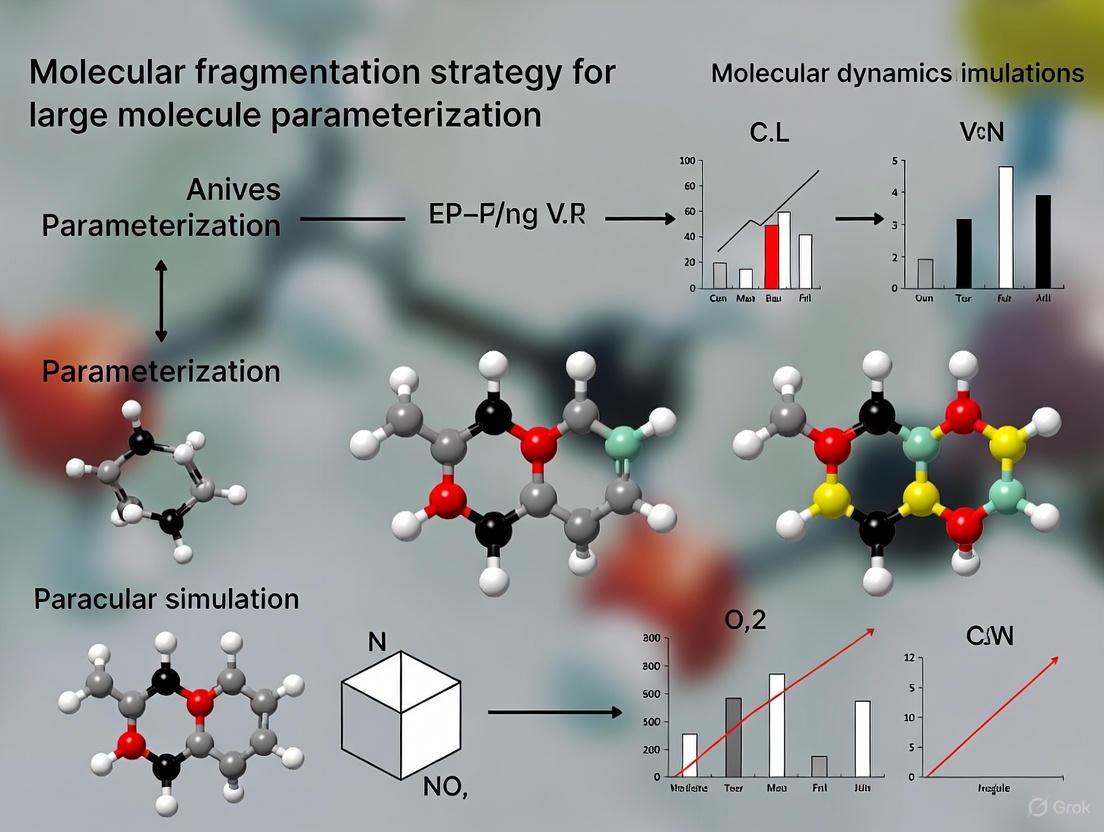
Advanced Molecular Fragmentation Strategies for Large Molecule Parameterization in Drug Discovery
Parameterizing large molecules for accurate molecular dynamics simulations is a central challenge in computational drug discovery.
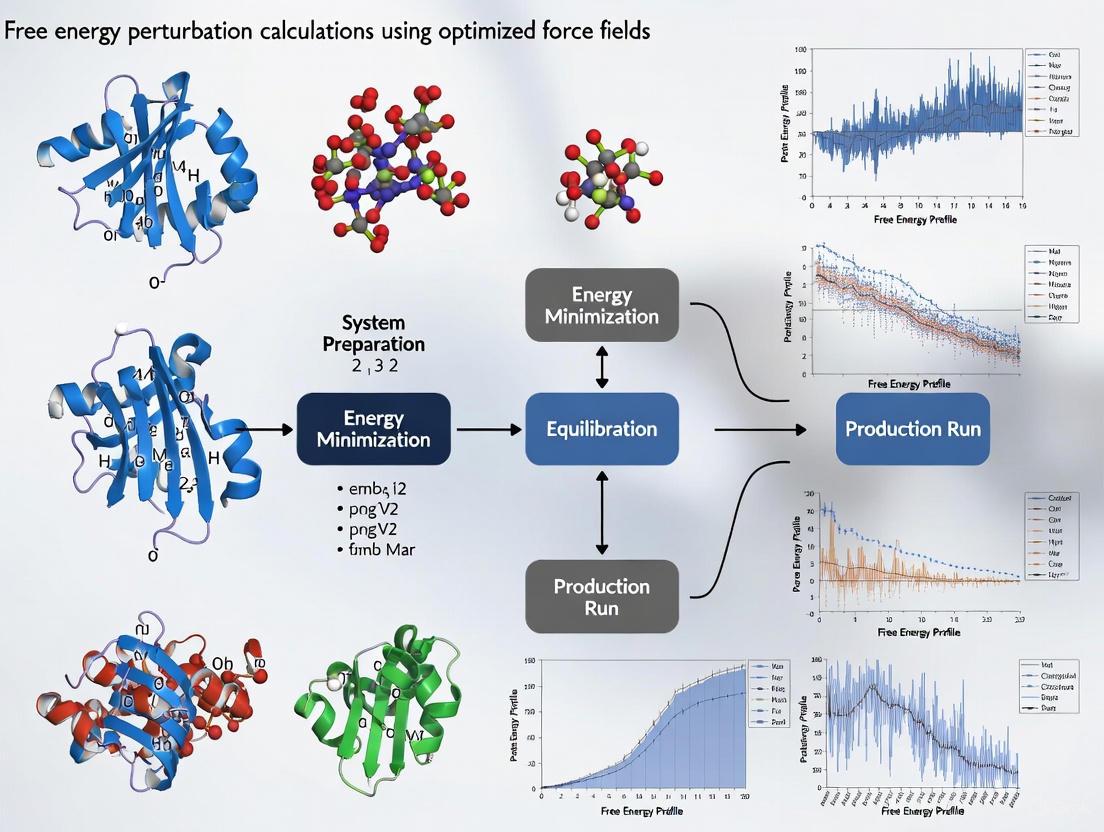
Optimizing Force Fields for Accurate Free Energy Perturbation Calculations in Drug Discovery
This article provides a comprehensive guide for researchers and drug development professionals on setting up and optimizing Free Energy Perturbation (FEP) calculations, a critical tool for predicting ligand binding affinities.
A Comprehensive Guide to RESP Charge Derivation: Protocols for Accurate Biomolecular Force Fields
This article provides a complete guide to the Restrained Electrostatic Potential (RESP) charge derivation protocol, a cornerstone of modern molecular mechanics force fields.
From Quantum Mechanics to Force Fields: A Comprehensive Guide to Parameter Conversion for Drug Discovery
This article provides a comprehensive overview of modern methodologies for converting quantum mechanical (QM) data into accurate molecular mechanics (MM) force field parameters, a critical process for reliable molecular dynamics...
Automated Force Field Optimization with Bayesian Inference: A Robust Workflow for Biomolecular Modeling and Drug Discovery
This article provides a comprehensive guide to automated force field parameter optimization using Bayesian inference, a transformative approach for computational researchers and drug development professionals.
Force Field Drift in MD Simulations: Causes, Solutions, and Validation for Biomedical Research
Long-timescale Molecular Dynamics (MD) simulations are pivotal for drug discovery and understanding biomolecular mechanisms, but their predictive power is often limited by force field drift—a gradual deviation from accurate physical...
Beyond the Table: The Critical Limitations of Traditional Look-up Table Force Fields and the Rise of Machine Learning
This article provides a comprehensive analysis of the limitations inherent in traditional look-up table-based force fields, a longstanding cornerstone of molecular dynamics simulations in drug development and biomolecular research.
Overcoming the Transferability Challenge: Force Field Parameterization Across Expansive Chemical Space in Drug Discovery
The accuracy of molecular dynamics simulations in drug discovery critically depends on the transferability of force field parameters across the vast and diverse landscape of drug-like molecules.