Research Articles

Mastering REMD Equilibration: A Practical Guide for Enhanced Sampling in Biomedical Research
This guide provides a comprehensive framework for achieving proper equilibration in Replica Exchange Molecular Dynamics (REMD), a critical enhanced sampling technique for studying complex biomolecular processes like protein folding and...
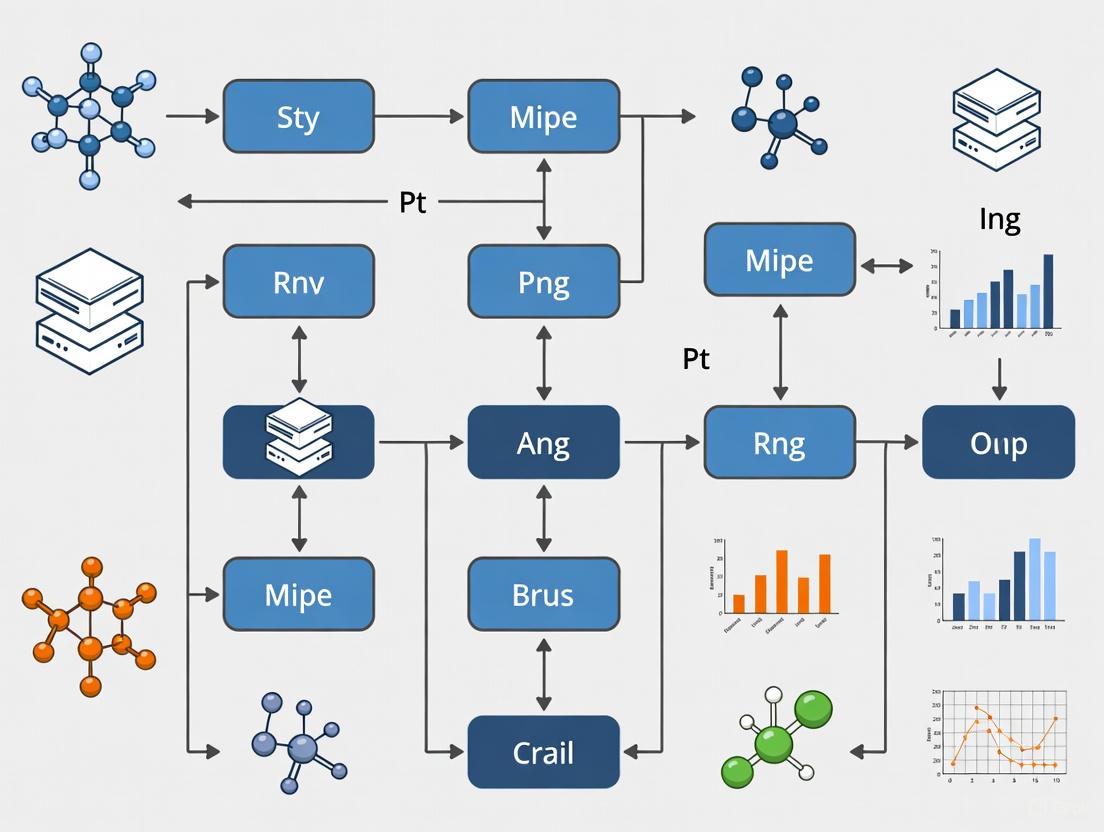
Strategies for Reducing Computational Cost in Long Molecular Dynamics Simulations
Long-timescale Molecular Dynamics (MD) simulations are pivotal for studying biomolecular processes but are often prohibitively expensive.
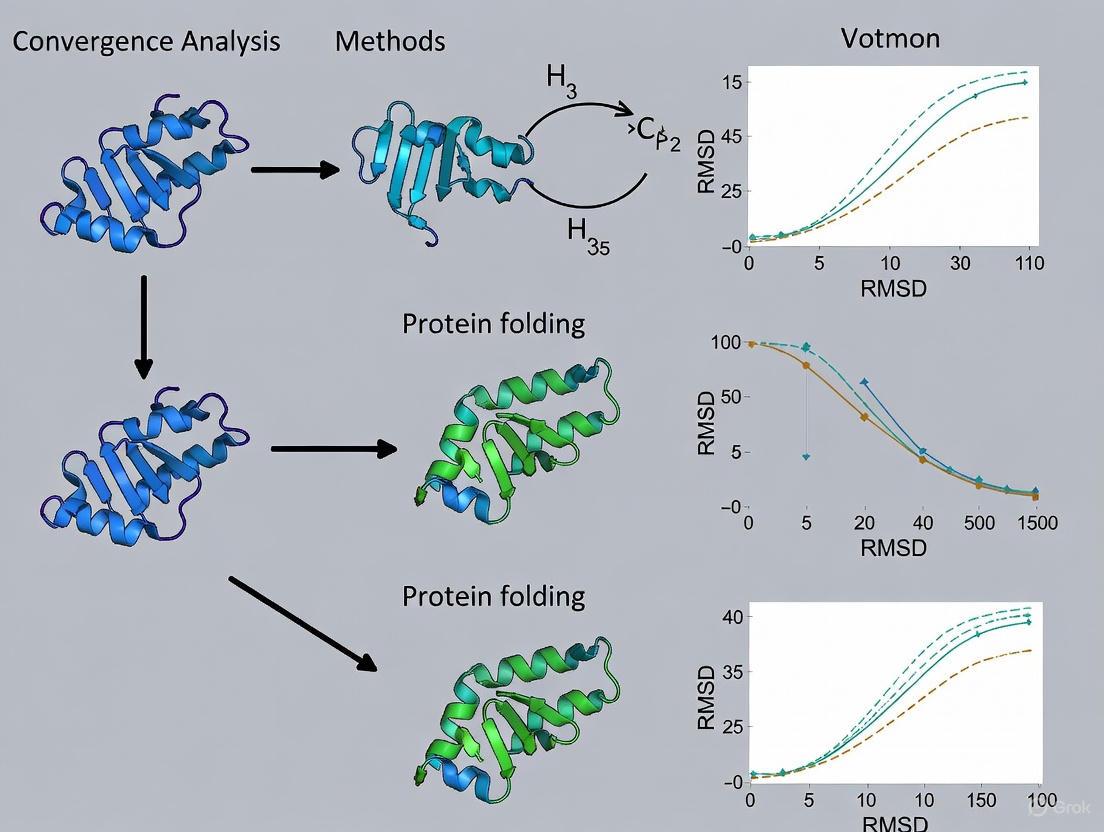
Convergence Analysis in Protein Folding: A Guide to Validating Trajectories from MD Simulations to Machine Learning
This article provides a comprehensive guide to convergence analysis for protein folding trajectories, a critical step for ensuring the reliability of computational studies.
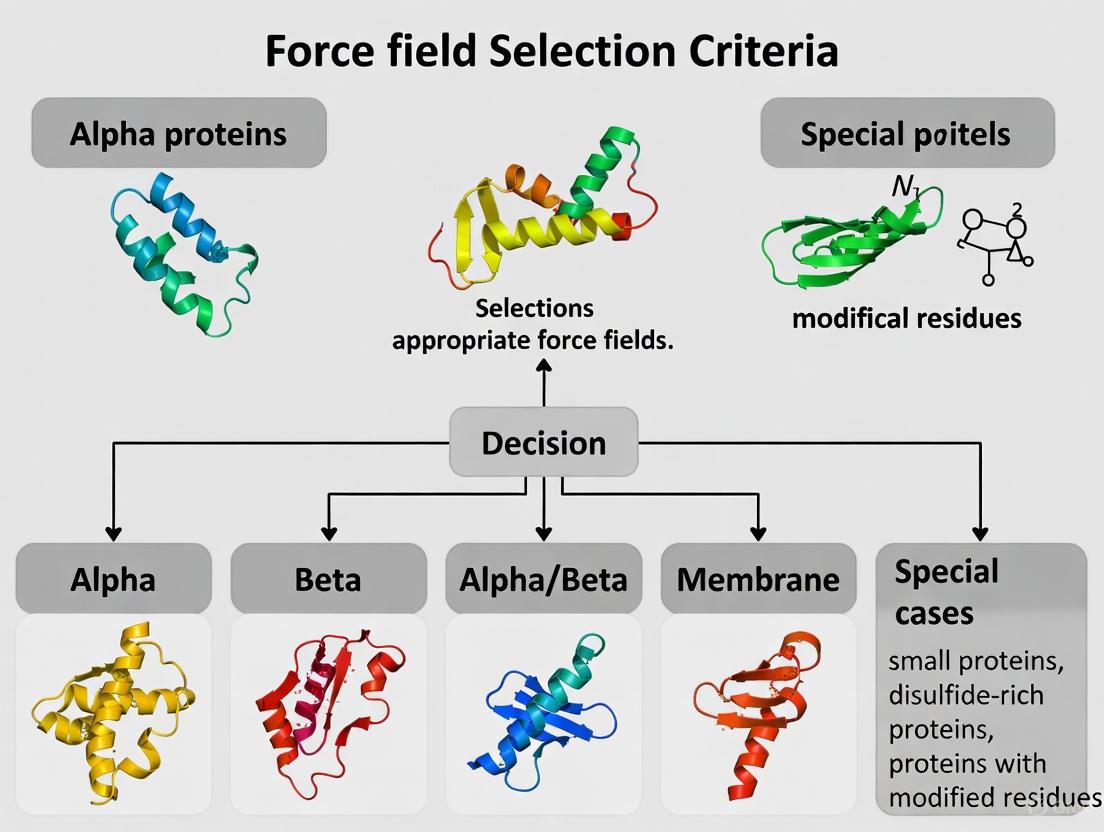
A Practical Guide to Force Field Selection for Protein Simulations: From Foundations to Future-Proofing
Accurate molecular dynamics simulations are paramount for advancing research in drug design and understanding protein function, yet their predictive power is fundamentally limited by the choice of force field.
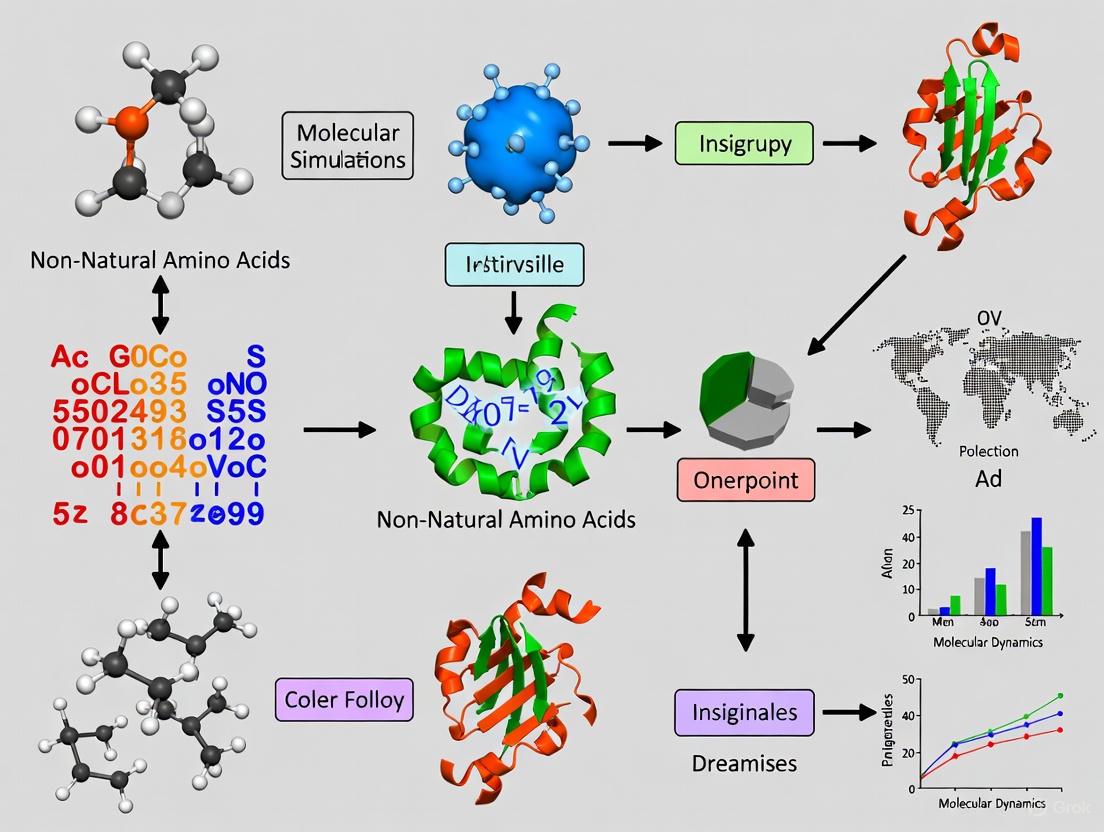
Navigating Non-Natural Amino Acids in Molecular Dynamics Folding Simulations: A Guide for Therapeutic Peptide Design
The incorporation of non-canonical amino acids (ncAAs) is a powerful strategy to enhance the stability, permeability, and binding affinity of therapeutic peptides.
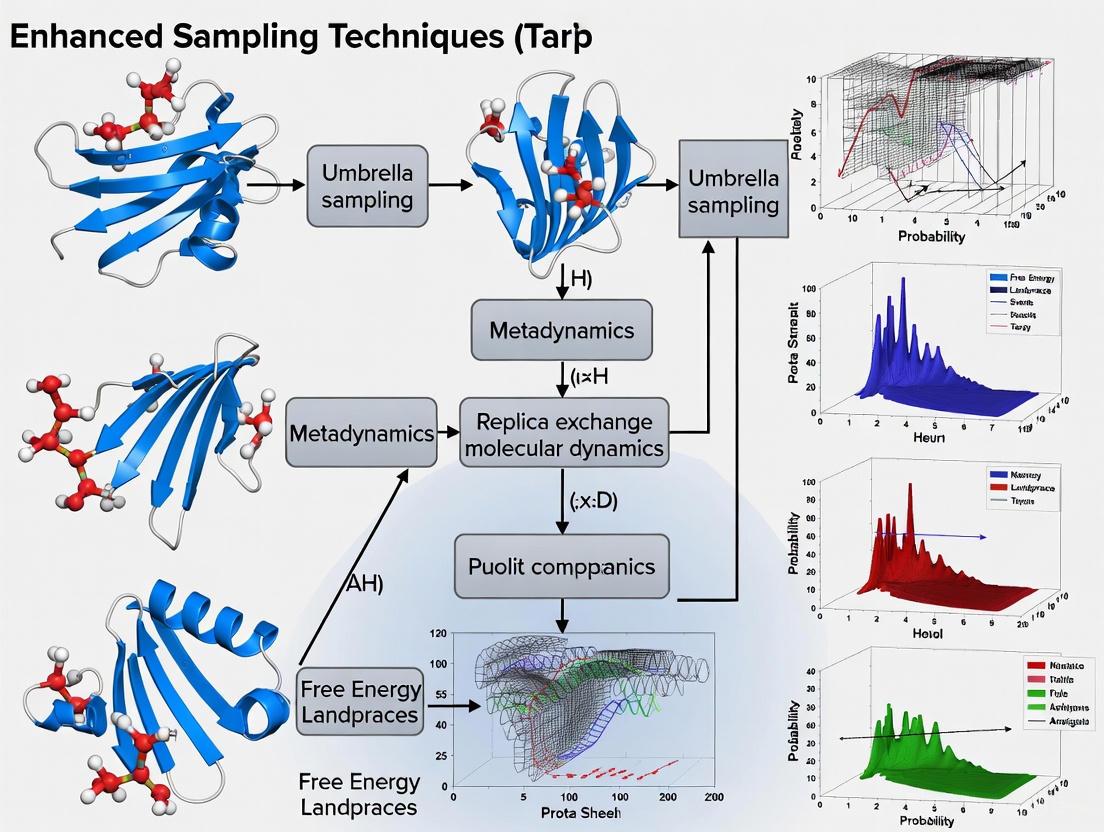
Breaking the Timescale Barrier: Enhanced Sampling Techniques for Simulating Rare Events in Protein Folding
This article provides a comprehensive overview of advanced computational methods designed to overcome the timescale limitations of molecular dynamics simulations in studying rare protein folding events.
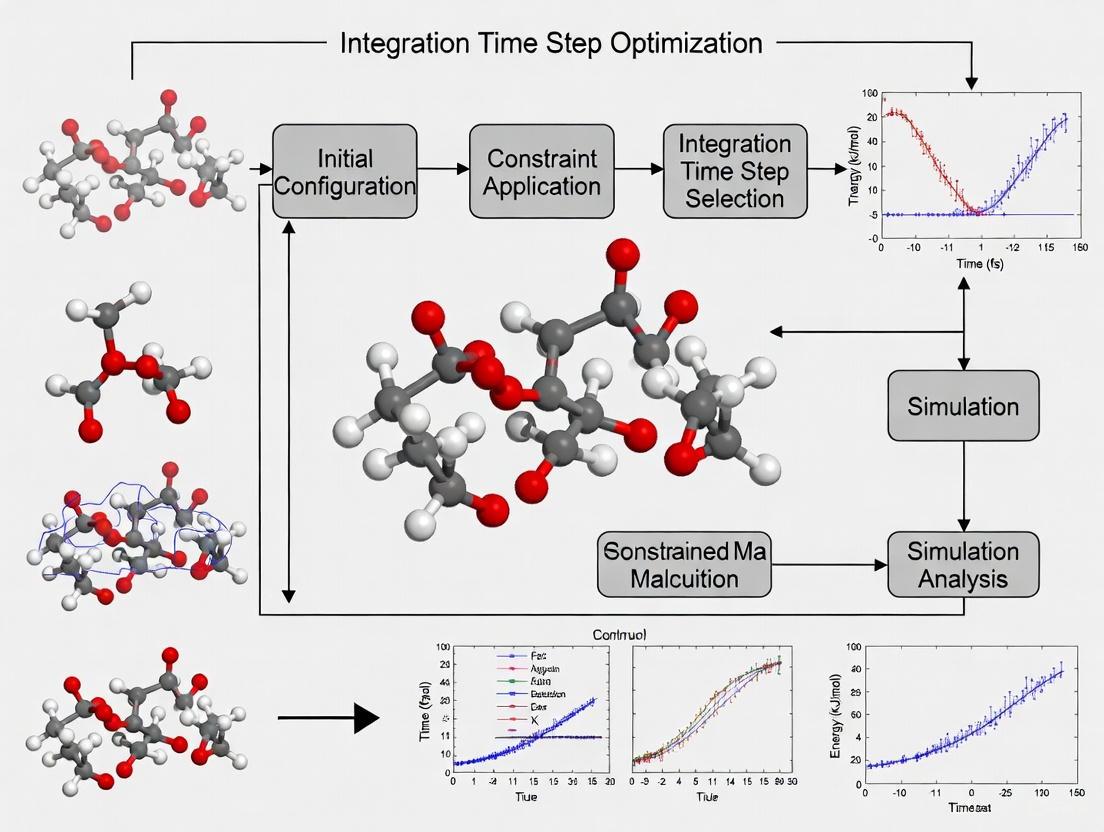
Optimizing Integration Time Steps in Constrained MD Simulations: A Guide for Biomedical Researchers
This article provides a comprehensive guide for researchers and drug development professionals on optimizing integration time steps in constrained Molecular Dynamics (MD) simulations.
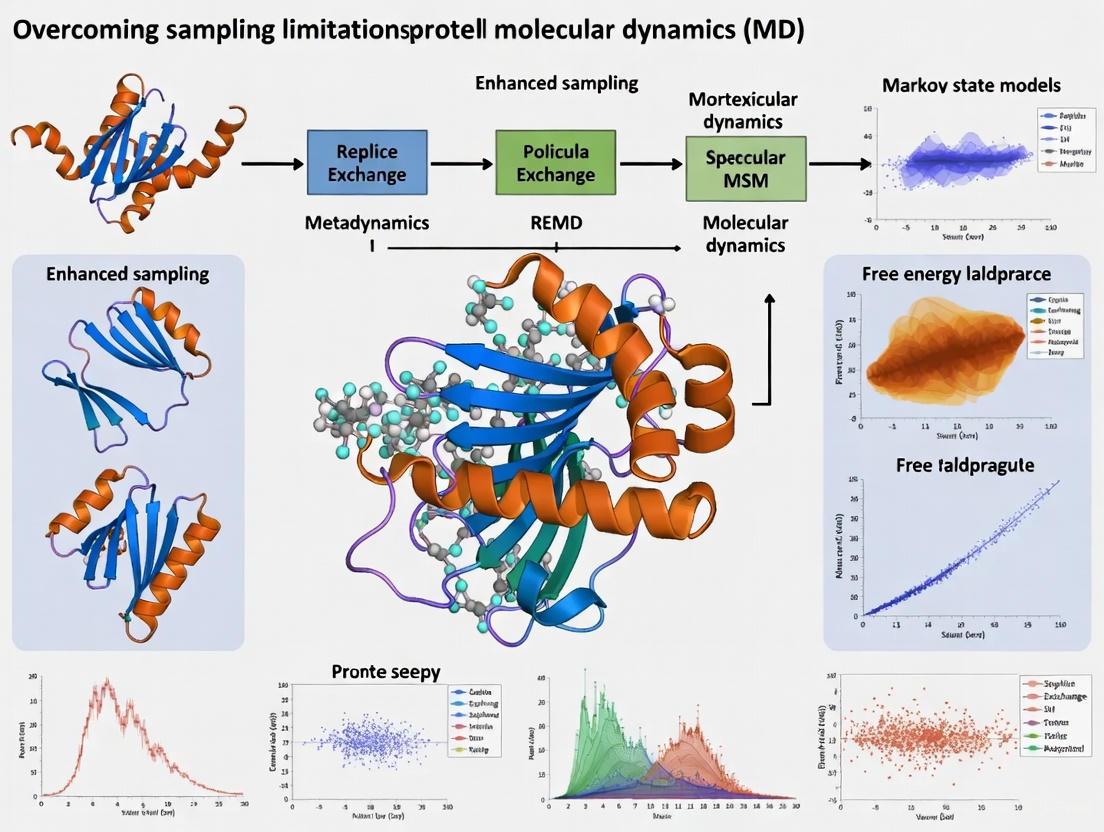
Beyond the Timescale Barrier: Modern Strategies to Overcome Sampling Limits in Small Protein Simulations
Molecular dynamics (MD) simulations are a cornerstone of structural biology and drug discovery, yet their utility is fundamentally constrained by the limited timescales accessible for sampling the conformational landscapes of...
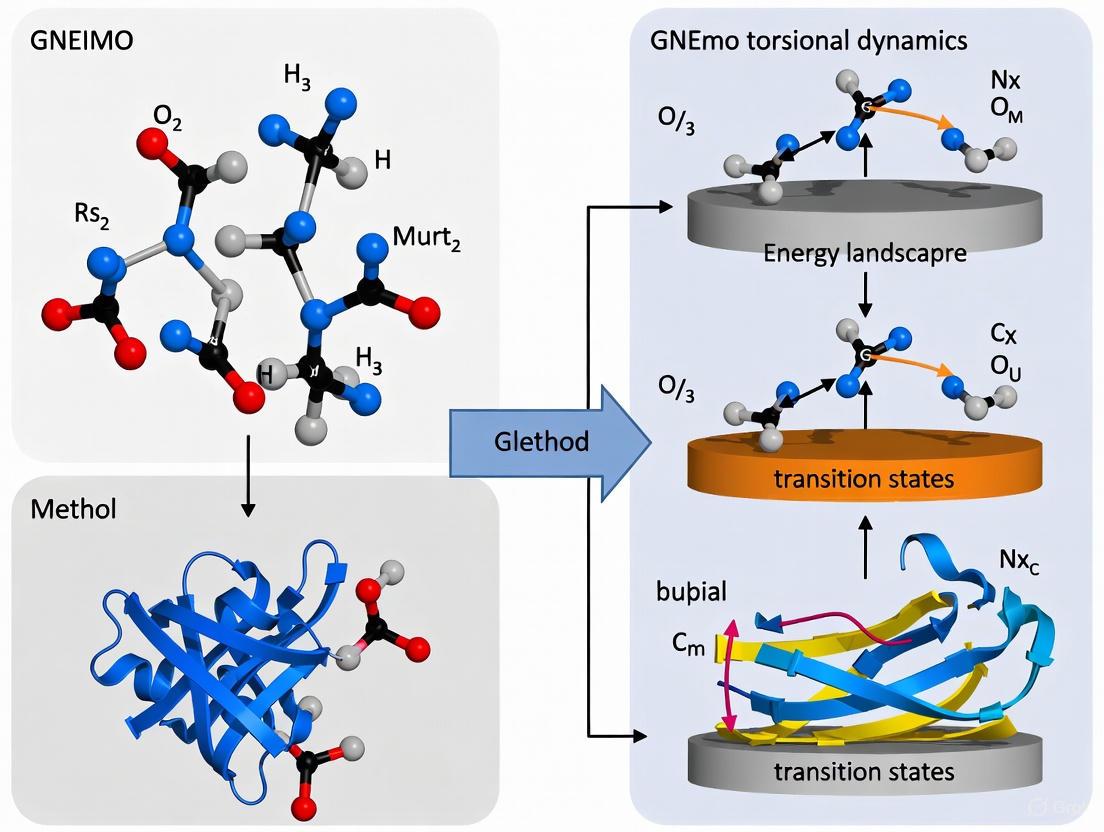
GNEIMO Method: Revolutionizing Protein Folding and Refinement with Torsional Dynamics
This article explores the Generalized Newton-Euler Inverse Mass Operator (GNEIMO) method, an advanced internal coordinate molecular dynamics (ICMD) technique transforming the study of protein folding and structure refinement.
Machine Learning Revolution: Next-Gen Coarse-Grained MD Parameters for Small Protein Folding
This comprehensive review explores the transformative impact of machine learning and advanced parameterization methods on coarse-grained molecular dynamics (CGMD) for small protein folding studies.